Oxidation
A continuing theme of our group's research is the selective oxidation of alkenes, and we have recently described the development of a procedure for the oxidative cyclisation of 1,5-dienes using catalytic (5 mol%) OsO4 under acidic conditions. This reaction is particularly powerful because it involves the stereospecific, suprafacial addition of two oxygen atoms across each of the two alkenes in 1 (scheme 1), coupled with the stereoselective formation of a cis-substituted tetrahydrofuran ring 2. Here we demonstrated that a range of mono, di and trisubstituted alkenes of either cis or trans geometry cyclise in good yields to give exclusively cis-tetrahydrofurans as single stereoisomers in each case. Furthermore, diene 3 cyclised under these conditions to give the THF 4, which was then transformed via a short sequence into the natural product D-chitaric acid.
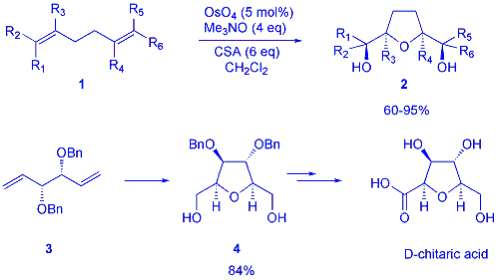
Attempts to induce asymmetric induction in this reaction failed, presumably owing to the low pH of the reaction. However, we have now shown that vicinal diols derived from 1,5-dienes can undergo cyclisation in a regio- and stereoselective manner to give cis-THFs under conditions comparable to diene cyclisation (scheme 2). The presence of a sacrificial alkene now greatly improves the yield of the reaction, as it allows the formation of Os (VI) in situ from Os (VIII) by a dihydroxylation reaction and thus acts to increase the amount of active Os (VI) catalyst in the reaction mixture. In this manner the diol 5 was transformed into the cis-THF 6 which was rapidly advanced into a known precursor to the annonaceous acetogenin (+)-cis-solamin.
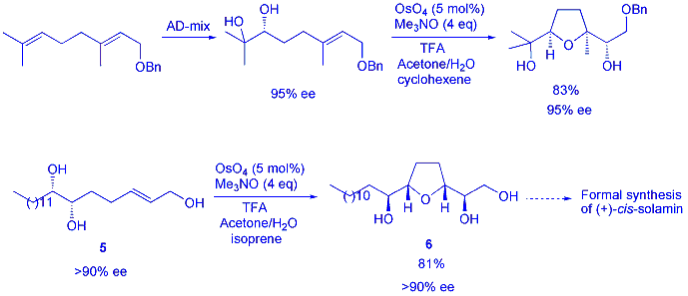
In a further advance to this chemistry we have also reported a double oxidative cyclisation en route to the first synthesis of the annonaceous acetogenin cis-sylvaticin. The protected tetrol 7 was obtained through a number of steps including double asymmetric dihydroxylation and a Wittig reaction to install the left-hand side chain (scheme 3). Subjection of this material to our oxidative cyclisation conditions (this time using trans-cinnamic acid as the sacrificial alkene to aid the workup procedure) served to excise both dimethyl acetal protecting groups and give the bis-THF 8 as a single, enantiopure diastereomer. This was then rapidly converted into the natural product, in the first asymmetric synthesis of cis-sylvaticin in only 13 linear steps and 19 chemical operations.
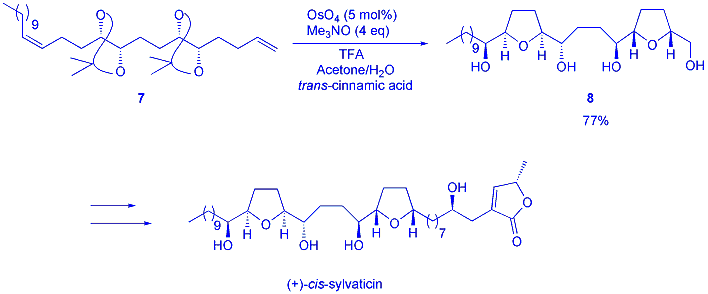
With the knowledge that diols can condense with Os (VI) species to give THFs, this raised the possibility of incorporating different intitatior groups for the oxidative cyclisation. Thus, we have shown that amino-alcohols and hydroxy-amides are efficient substrates for this reaction (scheme 4). Crucially, amino-alcohols can now give pyrrolidines as well as THFs in very high yields and with low catalyst loadings. In addition, enantiopure starting materials have been shown to give enantiopure heterocyclic products.
